

La méthode SAGE (Serial Analysis of genes expression)
Point scientifique
|
Principes
de la méthode SAGE
|
||
|
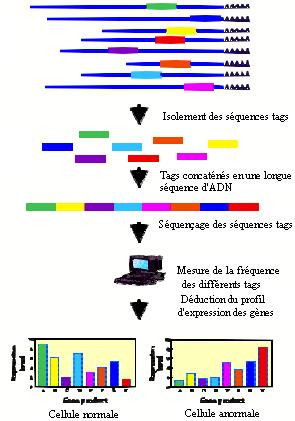
La méthode SAGE est un moyen d'étudier le transcriptome d'une cellule. La méthode SAGE, consiste à réaliser un inventaire des transcrits au moyen de courts fragments d'ADNc de 9 à 14 pb appelés sequence tags. Dans un premier temps, un pool d'ADNc est synthétisé à partir des ARNm d'un échantillon (cellules, tissus, organes...). Un traitement enzymatique par des enzymes de restriction permet ensuite d'isoler à partir des extrémités 3' de chacune des molécules d'ADNc, une courte séquence unique : le tag dont la taille est suffisante pour pouvoir identifier le gène dont elle dérive. Tous les tag (jusqu'à une cinquantaine) sont ensuite concaténés pour former un fragment d'ADNc qui puisse être manipulé et finalement séquencé. L'ensemble de ces tags (formant les concatémères) est le reflet qualitatif et quantitatif de chaque gène exprimé. Un traitement informatique des données permet d'une part de classer les séquences des fragments obtenus en fonction du nombre de fois où ils ont été observés et d'autre part de rechercher, dans les banques internationales, le gène (connu ou inconnu) auquel le tag correspond. |
||
|
Limites
de la méthode
|
||
|
Cette technique n'est pas adaptée à l'étude de nombreux échantillons et de transcrits rares. De plus, elle requiert l'existence de bases de données de séquences génomiques pour les espèces étudiées : on ne peut appréhender l'étude des transcriptomes cellulaires par l'analyse des ADN complémentaires que si l'on dispose de séquences complètes de gènes, ou bien des signatures de gènes (petites séquences du gène appelées EST). La méthode SAGE a déjà fait l'objet d'applications chez l'homme, chez la levure et chez les plantes. Le problème des tags est leur petite taille, et donc leur probabilite élevée d'etre trouve par hasard dans le génome. Un tag a une probablite de 1 sur N puissance 4 d'être présent aléatoirement dans le génome (N représente le nombre de base du tag). Une autre limitation de la technique est le taux d'erreur lors du sequençage. Si une erreur apparait durant le séquençage le tag sera erroné et il sera impossible de prédire sa localisation.
|
{kind=link}